Overview
The gDRcore is part of the gDR suite. The
package provides a set of tools to process and analyze drug response
data.
Introduction
Data model
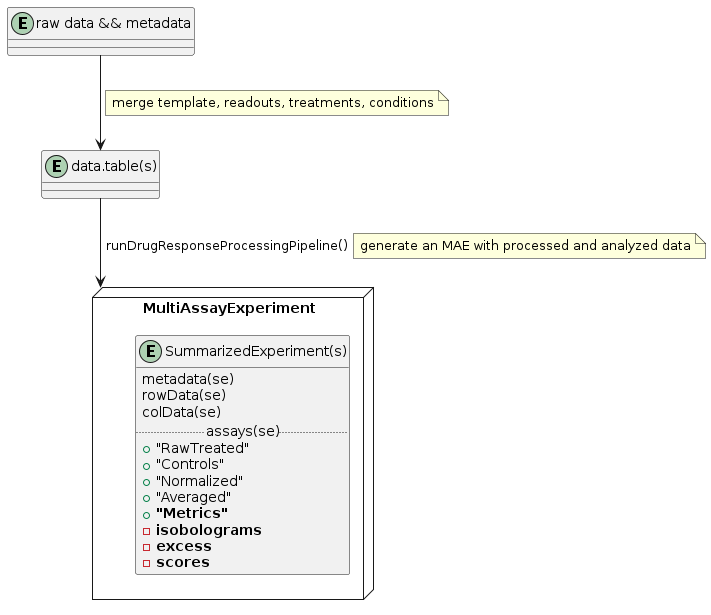
The data model is built on the MultiAssayExperiment (MAE) structure. Within an MAE, each SummarizedExperiment (SE) contains a different experiment type (e.g., single-agent or combination treatment). Columns of the MAE are defined by the cell lines and any modifications to them and are shared with the SEs. Rows are defined by the treatments (e.g., drugs, perturbations) and are specific to each SE. Assays of the SE are the different levels of data processing (raw, control, normalized, averaged data, as well as metrics). Each nested element of the assays of the SEs comprises the series themselves as a table (data.table in practice). Although not all elements need to have a series or the same number of elements, the attributes (columns of the table) should be consistent across the SE.
Drug processing
For drug response data, the input files need to be merged such that
each measurement (data) is associated with the correct metadata (cell
line properties and treatment definition). Metadata can be added with
the function cleanup_metadata if the right reference
databases are in place.
Required columns
To process the data through
runDrugResponseProcessingPipeline, the input data should
contain the required columns as well as optional columns.
For single-agent experiments, the required columns are: * Gnumber * DrugName * drug_moa * Concentration * clid * CellLineName * Tissue * ReferenceDivisionTime * parental_identifier * subtype * Duration * ReadoutValue
For combination experiments, additional required fields are: * Gnumber_2 * DrugName_2 * drug_moa_2 * Concentration_2
gDR supports the inclusion of any additional metadata in the long table for the pipeline. However, the most common supported by default are:
- Barcode (or Plate)
- BackgroundValue
- WellRow
- WellColumn
gDR pipeline
When the data and metadata are merged into a long table, the wrapper
function runDrugResponseProcessingPipeline can be used to
generate an MAE with processed and analyzed data.
 .
.
In practice, runDrugResponseProcessingPipeline performs
the following steps:
-
create_SE: Creates the structure of the MAE and the associated SEs by assigning metadata into the row and column attributes. The assignment is performed in the functionsplit_SE_components(see details below for the assumptions made when building SE structures).create_SEalso dispatches the raw data and controls into the right nested tables. Note that data may be duplicated between different SEs to make them self-contained. -
normalize_SE: Normalizes the raw data based on the control. Calculation of the GR value is based on a cell line division time provided by the reference database if no pre-treatment control is provided. If both pieces of information are missing, GR values cannot be calculated. Additional normalization can be added as new rows in the nested table. -
average_SE: Averages technical replicates that are stored in the same nested table. -
fit_SE: Fits the dose-response curves and calculates response metrics for each normalization type. -
fit_SE.combinations: Calculates synergy scores for drug combination data and, if the data is appropriate, fits along the two drugs and matrix-level metrics (e.g., isobolograms) are calculated. This is also performed for each normalization type independently.
 .
.
The functions used to process the data have parameters for specifying the names of the variables and assays. Additional parameters are available to personalize the processing steps, such as forcing the nesting (or not) of an attribute and specifying attributes that should be considered as technical replicates or not.
Use Cases
Data preprocessing
Please familiarize yourself with the gDRimport package,
which contains a variety of tools to prepare input data for
gDRcore.
This example is based on the artificial dataset called
data1 available within the gDRimport package.
gDR requires three types of data that should be used as the
raw input: Template, Manifest, and RawData. More information about these
three types of data can be found in our general documentation.
td <- gDRimport::get_test_data()The provided dataset needs to be merged into one
data.table object to be able to run the gDR pipeline. This
process can be done using two functions:
gDRimport::load_data() and
gDRcore::merge_data().
Running gDR pipeline
We provide an all-in-one function that splits data into appropriate
data types, creates the SummarizedExperiment object for each data type,
splits data into treatment and control assays, normalizes, averages,
calculates gDR metrics, and finally, creates the MultiAssayExperiment
object. This function is called
runDrugResponseProcessingPipeline.
mae <- runDrugResponseProcessingPipeline(input_df)
mae
#> A MultiAssayExperiment object of 2 listed
#> experiments with user-defined names and respective classes.
#> Containing an ExperimentList class object of length 2:
#> [1] combination: SummarizedExperiment with 2 rows and 6 columns
#> [2] single-agent: SummarizedExperiment with 3 rows and 6 columns
#> Functionality:
#> experiments() - obtain the ExperimentList instance
#> colData() - the primary/phenotype DataFrame
#> sampleMap() - the sample coordination DataFrame
#> `$`, `[`, `[[` - extract colData columns, subset, or experiment
#> *Format() - convert into a long or wide DataFrame
#> assays() - convert ExperimentList to a SimpleList of matrices
#> exportClass() - save data to flat filesAnd we can subset the MultiAssayExperiment to receive the SummarizedExperiment specific to any data type, e.g.
mae[["single-agent"]]
#> class: SummarizedExperiment
#> dim: 3 6
#> metadata(5): identifiers experiment_metadata Keys fit_parameters
#> .internal
#> assays(5): RawTreated Controls Normalized Averaged Metrics
#> rownames(3): G00002_drug_002_moa_A_168 G00004_drug_004_moa_A_168
#> G00011_drug_011_moa_B_168
#> rowData names(4): Gnumber DrugName drug_moa Duration
#> colnames(6): CL00011_cellline_BA_breast_cellline_BA_unknown_26
#> CL00012_cellline_CA_breast_cellline_CA_unknown_30 ...
#> CL00015_cellline_FA_breast_cellline_FA_unknown_42
#> CL00018_cellline_IB_breast_cellline_IB_unknown_54
#> colData names(6): clid CellLineName ... subtype ReferenceDivisionTimeData extraction
Extraction of the data from either MultiAssayExperiment
or SummarizedExperiment objects into more user-friendly
structures, as well as other data transformations, can be done using
gDRutils. We encourage reading the gDRutils
vignette to familiarize yourself with these functionalities.
SessionInfo
sessionInfo()
#> R version 4.6.1 (2026-06-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] gDRcore_1.11.5 gDRtestData_1.10.0 BiocStyle_2.40.0
#>
#> loaded via a namespace (and not attached):
#> [1] farver_2.1.2 fastmap_1.2.0
#> [3] BumpyMatrix_1.20.0 TH.data_1.1-5
#> [5] digest_0.6.39 lifecycle_1.0.5
#> [7] gDRutils_1.10.0 survival_3.8-6
#> [9] magrittr_2.0.5 compiler_4.6.1
#> [11] rlang_1.2.0 sass_0.4.10
#> [13] drc_3.0-1 tools_4.6.1
#> [15] plotrix_3.8-14 yaml_2.3.12
#> [17] data.table_1.18.4 knitr_1.51
#> [19] lambda.r_1.2.4 S4Arrays_1.12.0
#> [21] htmlwidgets_1.6.4 DelayedArray_0.38.2
#> [23] RColorBrewer_1.1-3 abind_1.4-8
#> [25] multcomp_1.4-30 BiocParallel_1.46.0
#> [27] purrr_1.2.2 BiocGenerics_0.58.1
#> [29] desc_1.4.3 grid_4.6.1
#> [31] stats4_4.6.1 scales_1.4.0
#> [33] MASS_7.3-65 gtools_3.9.5
#> [35] MultiAssayExperiment_1.38.0 SummarizedExperiment_1.42.0
#> [37] cli_3.6.6 mvtnorm_1.4-1
#> [39] rmarkdown_2.31 ragg_1.5.2
#> [41] generics_0.1.4 otel_0.2.0
#> [43] readxl_1.5.0 cachem_1.1.0
#> [45] stringr_1.6.0 splines_4.6.1
#> [47] gDRimport_1.10.0 assertthat_0.2.1
#> [49] parallel_4.6.1 formatR_1.14
#> [51] BiocManager_1.30.27 cellranger_1.1.0
#> [53] XVector_0.52.0 matrixStats_1.5.0
#> [55] vctrs_0.7.3 Matrix_1.7-5
#> [57] sandwich_3.1-1 jsonlite_2.0.0
#> [59] carData_3.0-6 bookdown_0.47
#> [61] car_3.1-5 IRanges_2.46.0
#> [63] S4Vectors_0.50.1 Formula_1.2-5
#> [65] systemfonts_1.3.2 testthat_3.3.2
#> [67] jquerylib_0.1.4 rematch_2.0.0
#> [69] glue_1.8.1 pkgdown_2.2.0
#> [71] codetools_0.2-20 stringi_1.8.7
#> [73] futile.logger_1.4.9 GenomicRanges_1.64.0
#> [75] tibble_3.3.1 pillar_1.11.1
#> [77] htmltools_0.5.9 Seqinfo_1.2.0
#> [79] brio_1.1.5 R6_2.6.1
#> [81] textshaping_1.0.5 evaluate_1.0.5
#> [83] lattice_0.22-9 Biobase_2.72.0
#> [85] futile.options_1.0.1 backports_1.5.1
#> [87] bslib_0.11.0 SparseArray_1.12.2
#> [89] checkmate_2.3.4 xfun_0.59
#> [91] fs_2.1.0 MatrixGenerics_1.24.0
#> [93] zoo_1.8-15 pkgconfig_2.0.3